近日,国际蛋白质设计竞赛GEMxAdap-ty RBX1 Binder Design Competition 公布最终结果:「力文所」算法团队以全球第5名的成绩入围,也是国内唯一的团队。
据介绍,该赛事在全球累计收到11,784个设计方案,经严格筛选后对322个设计结果进行了实验验证,最终仅9个成功获得binder认定。值得关注的是,9个binder中只有两个具备:抑制E3泛素连接酶活性的设计结果。力文所则是其中之一,其设计结果能有效抑制表位功能。该项设计基于力文所团队自研的Pallatom2-binder 模型,采用全原子生成范式,结构序列一步输出,全程无任何后优化处理,且序列在已知蛋白数据库中无任何同源匹配(Seqldentity为0%)。
01
RBX1:蛋白质降解系统的"总控开关"
RBX1(Ring-box protein 1)是 E3 泛素连接酶 复合物的核心组件。E3 泛素连接酶是细胞内的"蛋白质降解机器":它识别特定的靶蛋白,给它们贴上泛素标签,标记为"待销毁",最终被蛋白酶体降解。清除错误蛋白、调控细胞周期、应对病毒感染——这些关键生命活动都离不开这套系统。
RBX1在E3复合物中扮演着连接"骨架"与"催化亚基"的关键桥梁角色。它的功能状态直接决定整个E3复合物能否正常工作。谁能精准调控RBX1,谁就能掌控蛋白质的降解命运。
本次 GEM × Adaptyv RBX1 Binder Design Competition 要求参赛者设计能特异性结合RBX1的蛋白质 binder。难点在于:binder 的结合位点必须落在RBX1的功能敏感区,才能真正干扰其生物学活性——而不仅仅是"粘上去"。
竞赛吸引了全球众多团队,累计提交11,784个设计,涵盖 BindCraft、BoltzGen、Proteina-Complexa、RFdiffusion等多种前沿方法。经严格筛选:对322个设计结果进行了实验验证,81%的整体表达率已属不易,但最终仅有9个成功获得 binder 认定。
02
对蛋白质锚点进行"双头锚定"
团队采用自研的 Pallatom2-binder 模型进行设计,核心策略是对蛋白质锚点进行"双头锚定"(粉色高亮区域标示了靶点的两个独立关键结合界面)。该策略一方面是能够将 binder 精确锁定在功能敏感区域,从而有效干扰 E3 复合物的正常催化活性,另一方面是可以稳定游离的loop结构,从而形成稳定的多点互作。
从竞赛官方公布的结合亲和力排名图可见,在全部 322 个与 RBX1 进行结合测试的蛋白质中,力文所的设计(noble-lion-fern)以约 1.9 × 10⁻⁷ M 的 KD 值稳定处于 binder 认定的阈值之上,位列第 5。
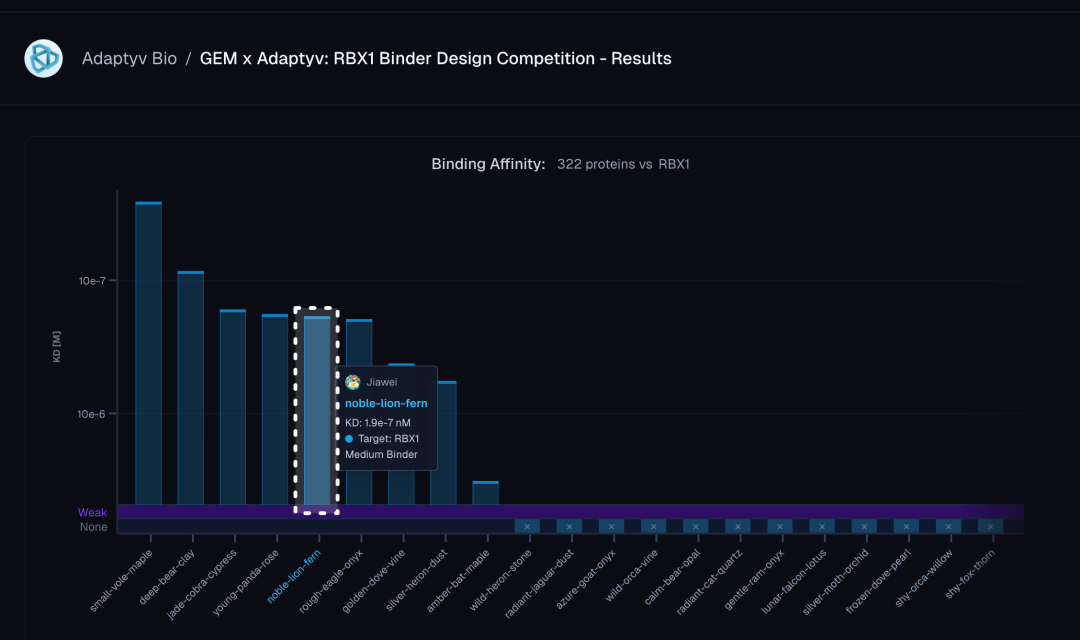
实验详情页进一步验证了这一结果:SPR 动力学曲线显示,在10 nM至1000 nM 的梯度浓度下,binder与RBX1 均呈现出典型的结合-解离特征,结合速率常数KON为 3.9 × 10⁴ M⁻¹s⁻¹,解离速率常数KOFF为 7.4 × 10⁻³ s⁻¹,计算所得KD 为 1.9 × 10⁻⁷ M,与三次独立重复的平均值高度一致。平台同步给出的ESMFold 结构预测图则展示了该binder的三维构象,整体折叠质量高。
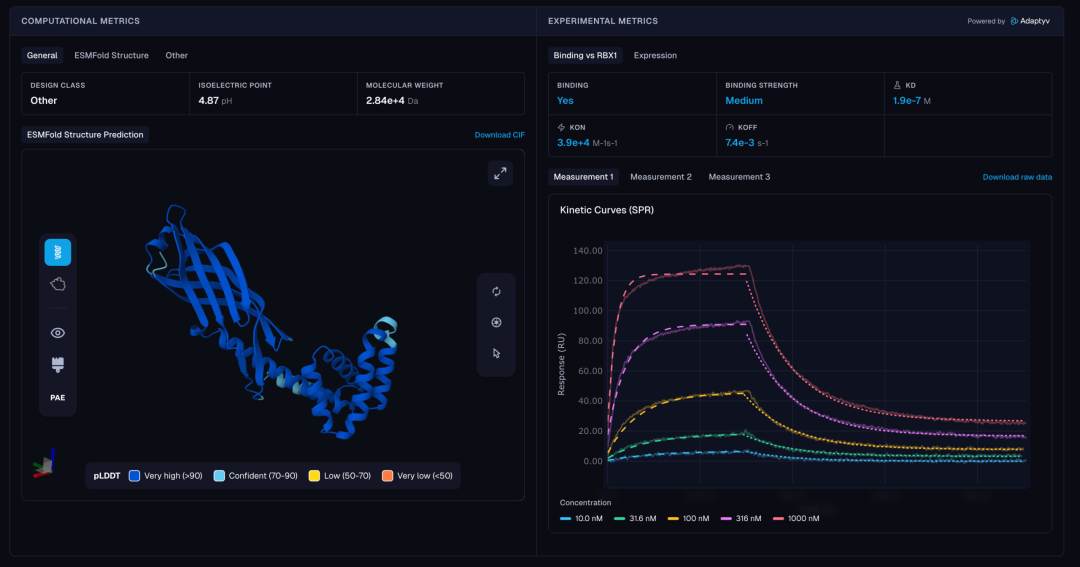
ESMFold 结构预测图
03
全原子生成:一步出结果
什么是"全原子生成"?
当前主流的蛋白质设计方法通常遵循"先生成骨架、再设计序列"的两步流程:第一步输出仅含主链原子的"骨架",第二步用序列设计工具为骨架匹配合适的氨基酸,第三步再预测序列的全原子结构,进行折叠验证。这个链条越长,信息损失和误差累积就越多——骨架生成时未考虑侧链的空间位阻,序列设计时未考虑侧链与靶点的精细相互作用,最终往往需要多轮迭代才能收敛到可用设计。
力文所使用自研的 Pallatom2-binder 模型采用的全原子生成则完全不同:它直接由靶点结构同步输出 binder 的完整三维构象,包括主链和全部侧链原子。这意味着从生成之初,每一个氨基酸的侧链构象都与主链折叠状态、与靶点的结合界面、与周围残基的空间关系在统一的框架内进行了协同优化,而非事后“补救”。
"一步出结果"的方法学优势
传统多步流程将"结构折叠"和"序列设计"割裂为两个独立问题,各自优化局部目标,难以保证全局最优。Pallatom2 将二者纳入统一的端到端框架:结构折叠为序列提供几何约束,序列选择反过来决定结构的能量景观,二者在生成过程中相互迭代、共同收敛。这种协同机制使得模型能够学习到传统分步方法难以捕捉的精细相互作用。
零后优化:原生能力的极限验证
团队未对 Pallatom2-binder 的输出进行任何后处理——无 ProteinMPNN 序列重设计、无 Rosetta 能量最小化。实验验证结果完全来源于模型的原生输出。
这意味着:Pallatom2 仅凭自身学习到的结构-序列规律,构象结合规律,便直接"直觉性"地生成了可结合、可表达、甚至可抑制功能的蛋白质。这一结果在方法学层面具有重要意义——它证明了端到端生成模型已具备不依赖物理势能面优化而独立输出高质量设计的能力。
全新序列,零同源
实验序列的SeqIdentity检测为0%,意味着该binder的序列在UniProt等数据库中无任何已知同源蛋白,是一个彻头彻尾的de novo设计。
传统方法本质上是在天然蛋白序列的基础上做"微整形",即使改动了部分残基,骨架序列仍与天然蛋白保持较高同源性。而 Pallatom2 作为端到端生成模型,学习的不是"哪些天然序列好用",而是蛋白质折叠和分子识别的深层物理规律。它生成的序列虽然与天然蛋白完全不同,但氨基酸的类型、位置、相互作用模式都具有物理化学约束的自洽性——这使得序列全新,却仍能正确折叠、稳定表达、并执行预期的结合功能。
04
端到端范式的可行性验证
传统蛋白质设计依赖"生成骨架 + 序列优化 + 能量最小化"的多步迭代流程,后优化往往是成败的关键。Pallatom2 在本次竞赛中以零后优化的方式直接产出功能性 binder,证明端到端生成模型已经具备了不依赖人工调参、不依赖物理势能面迭代而独立输出高质量设计的能力。这意味着蛋白质设计范式正在从"工具辅助人工"向"模型一步生成"发生根本性转变。
全链路设计能力的系统性验证
竞赛的严苛筛选机制(设计 → 表达 → 结合 → 功能)实际上构成了一条完整的"计算设计-实验验证-功能评估"能力检验链。力文所算法团队能够在这条链上全通,说明团队自研的算法模型已经可以无缝融入从靶点分析、模型生成到生物学功能验证的完整闭环内。
面向更广泛场景的应用潜力
基于验证后的端到端生成能力,团队的技术平台可以面向酶工程、抗体设计、信号调控蛋白、新型疫苗抗原等多个方向展开探索。特别是在功能性蛋白质设计这一高壁垒领域,端到端模型"一步到位"的能力将显著缩短从概念到验证的周期,为蛋白质药物和合成生物学工具的开发提供更高效的基础设施。
来源:力文所

©2021 磐霖资本保留所有权利 沪ICP备10037119号-1  沪公网安备 31011502019370号
沪公网安备 31011502019370号